Electrophilic Aromatic Substitution (EAS)
The question this page answers: how do the C=C bonds in aromatic rings react with electrophiles — and why do substituents already on the ring decide where the new group ends up?
Deeper reading: Clayden, Organic Chemistry 2e, ch. 21 (pp. 473–496) and ch. 24 (pp. 562–567) — see our chapter-by-chapter practice map for Clayden.
Why benzene doesn't react like an alkene
Why won't benzene add Br2 the way an alkene does?
The C=C π bonds in aromatic rings are far less reactive than ordinary alkenes because they are stabilized by aromatic delocalization.
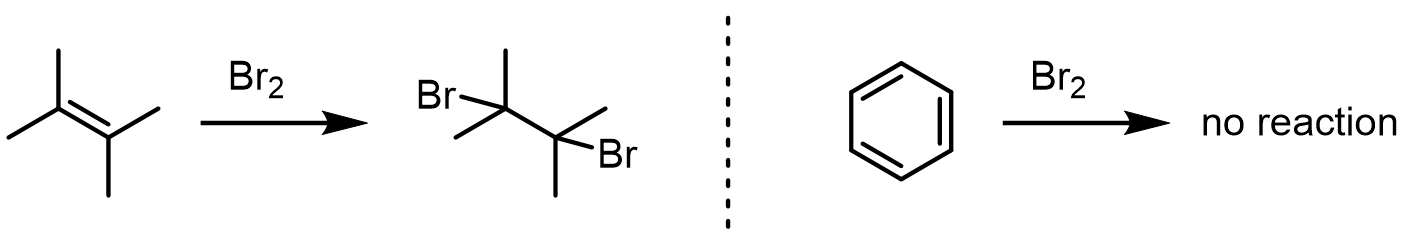
Losing aromaticity is expensive, so an aromatic π system will not attack a run-of-the-mill electrophile. To get a reaction, you need a much stronger electrophile — and that requirement is what defines this whole family of chemistry.
The five classic EAS reactions
What are the five classic EAS reactions?
Each named EAS reaction is just a different recipe for generating a strong electrophile.
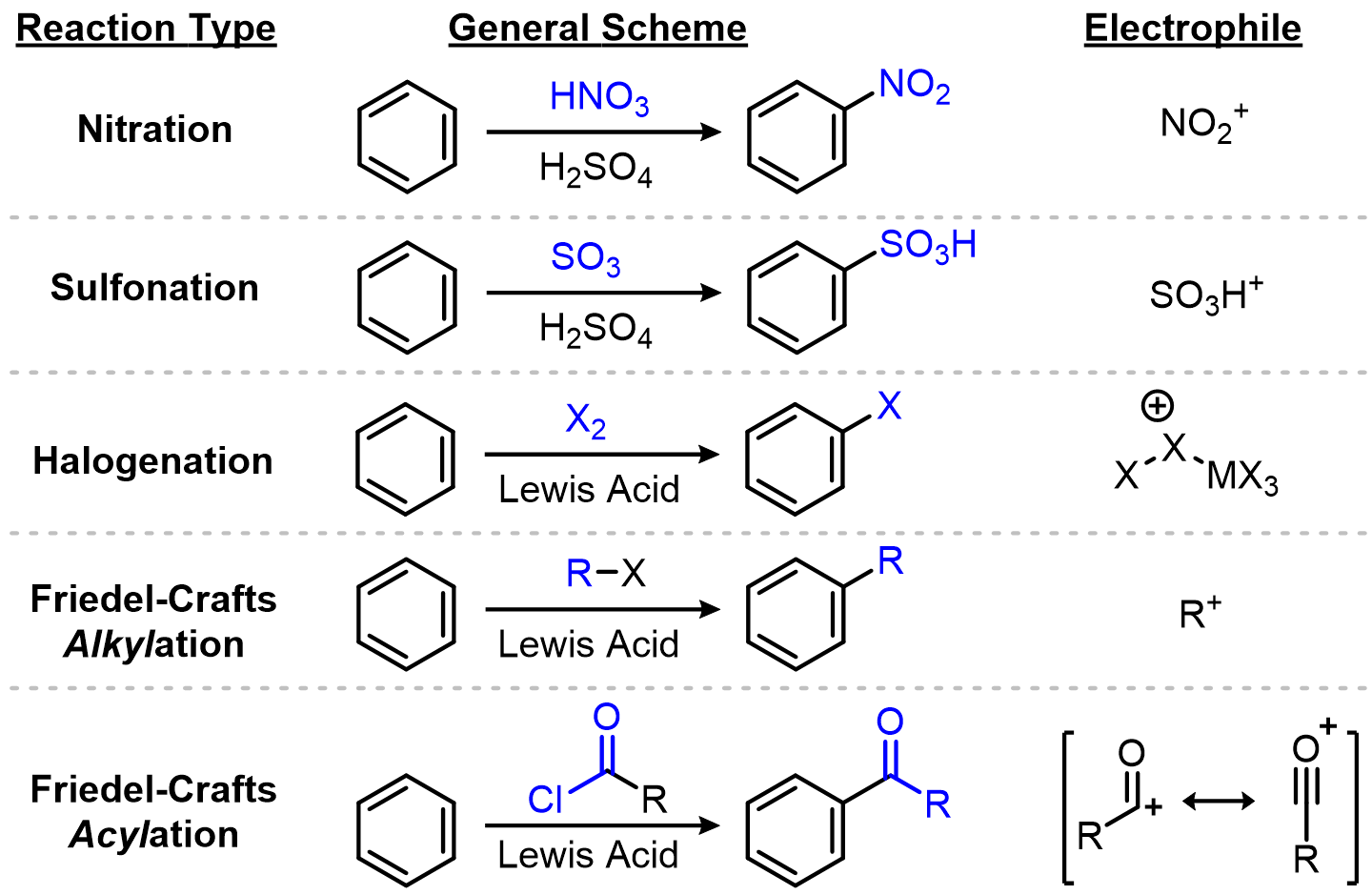
One mechanism to rule them all
What mechanism do all EAS reactions share?
Every EAS reaction has the same two steps: (1) addition of the electrophile to a C=C bond to form an arenium ion intermediate, then (2) a β-elimination that re-aromatizes the ring.
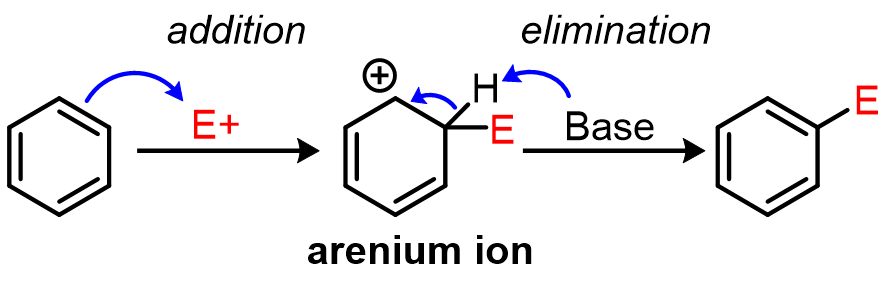
Notice what makes this a substitution overall: the ring temporarily gives up aromaticity in the addition step, then gets it back by kicking out a proton rather than adding a nucleophile. The arenium ion (also called a sigma complex or Wheland intermediate) is the key species — anything that stabilizes it speeds up the reaction.
Directing effects: where does the new group go?
Where does the new group end up — and why?
Groups already on the ring do two things: they direct the electrophile to particular positions, and they activate or deactivate the ring by donating or withdrawing electron density.
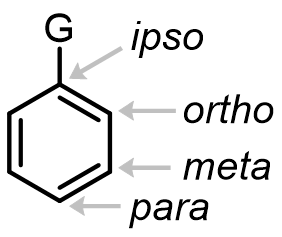
Directing groups come in exactly two flavors: ortho/para directors and meta directors. Here is nitration of anisole showing that –OMe is an ortho/para director:
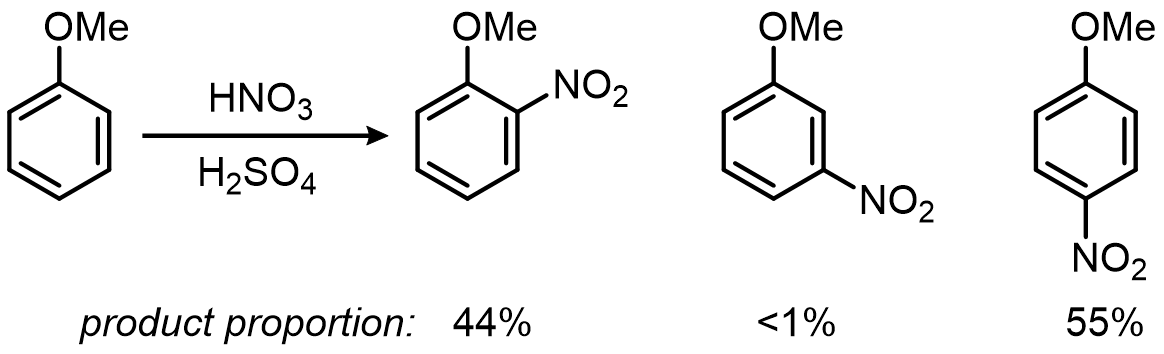
And nitration of nitrobenzene showing that –NO2 is a meta director:
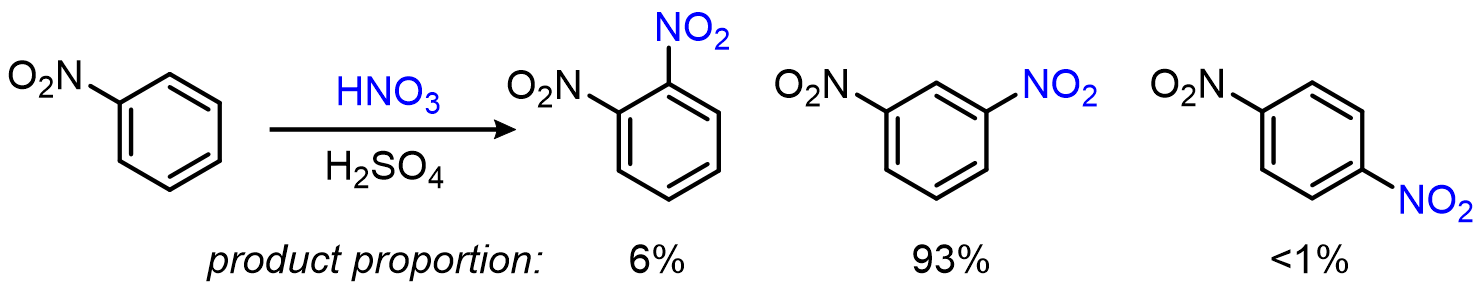
The rate side of the story: electron-donating groups (EDGs) increase electron density in the π system and make the ring more reactive; electron-withdrawing groups (EWGs) do the opposite.
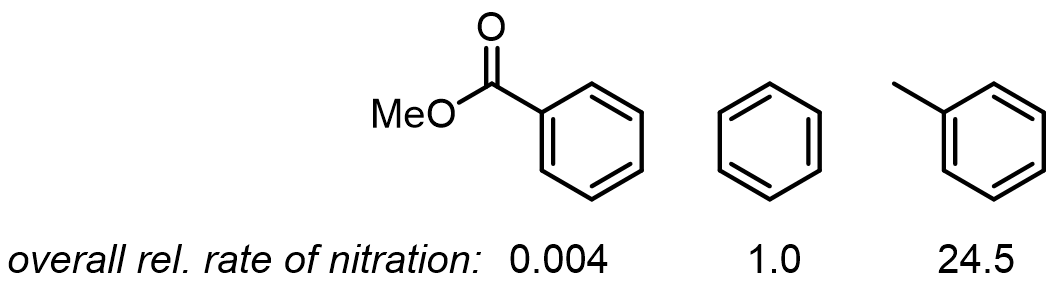
Taken together, the rules compress to three lines:
- EDGs activate the ring and direct ortho/para.
- Halogens deactivate the ring but still direct ortho/para (the classic exception).
- All other EWGs deactivate the ring and direct meta.
Multiple substituents: balancing the effects
What if two directing groups disagree?
With more than one group on the ring, sum the individual effects. When groups disagree, the reaction (1) avoids sterically crowded positions and (2) follows the group whose lone pair best stabilizes the arenium intermediate.
When the directors cooperate, the outcome is clean:
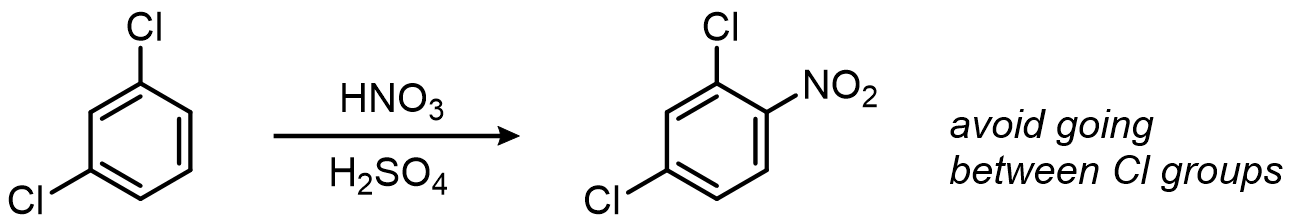
When they disagree, the stronger director wins:
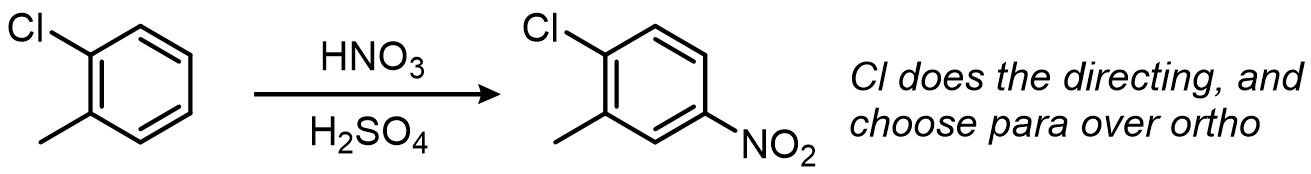
Friedel-Crafts reactions: powerful but tricky
Why does Friedel-Crafts alkylation misbehave — and what's the fix?
Friedel-Crafts reactions are prized because they make C–C bonds — but alkylation has two famous failure modes, and acylation is the workaround.
Problem one: alkylations go through carbocation-like intermediates, so they rearrange:
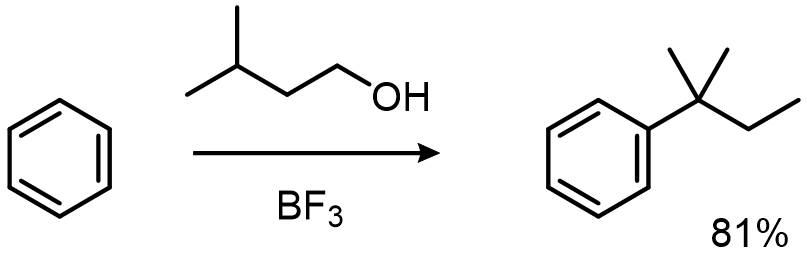
Problem two: each alkyl group you install activates the ring, so the product outcompetes the starting material and you over-alkylate:
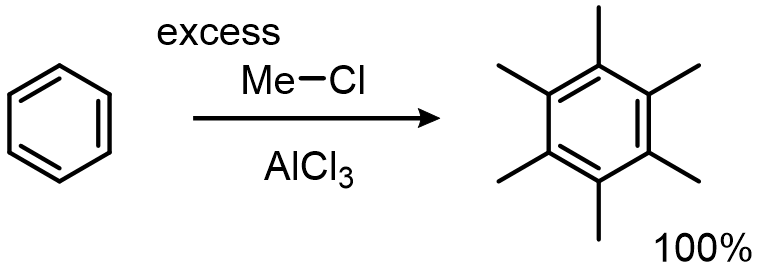
The fix for both: acylate instead, then reduce. The acylium ion doesn't rearrange, and the ketone product is deactivated toward further EAS:
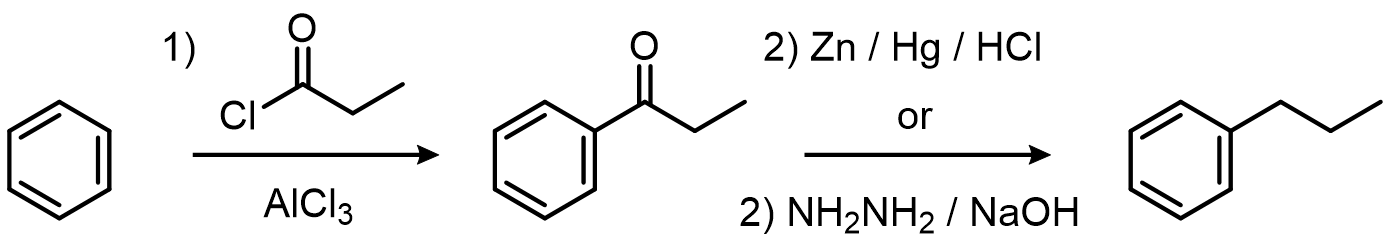
More Practice for this Topic
Reading about directing effects is one thing — predicting real products is another. Try it:
- Electrophilic aromatic substitution worksheet — EAS reactions curated from the chemical literature (answer key)
- Identify Aromaticity — interactive practice deciding whether rings are aromatic, antiaromatic, or nonaromatic
- Nucleophilic aromatic substitution worksheet — the complementary reaction family (answer key)
Spotted an error, or want a topic covered next? Let us know.